Call:
glm(formula = Malignant ~ Radius + Concave, family = "binomial",
data = cell)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -13.1320 1.4932 -8.795 < 2e-16 ***
Radius 2.7175 0.3663 7.418 1.19e-13 ***
Concave 3.3192 0.3545 9.362 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 751.44 on 568 degrees of freedom
Residual deviance: 224.02 on 566 degrees of freedom
AIC: 230.02
Number of Fisher Scoring iterations: 724-Classification Metrics
SDS 291
Prof. Baumer
December 1, 2025
Example: Malignant tunors
Identifying malignant tumors
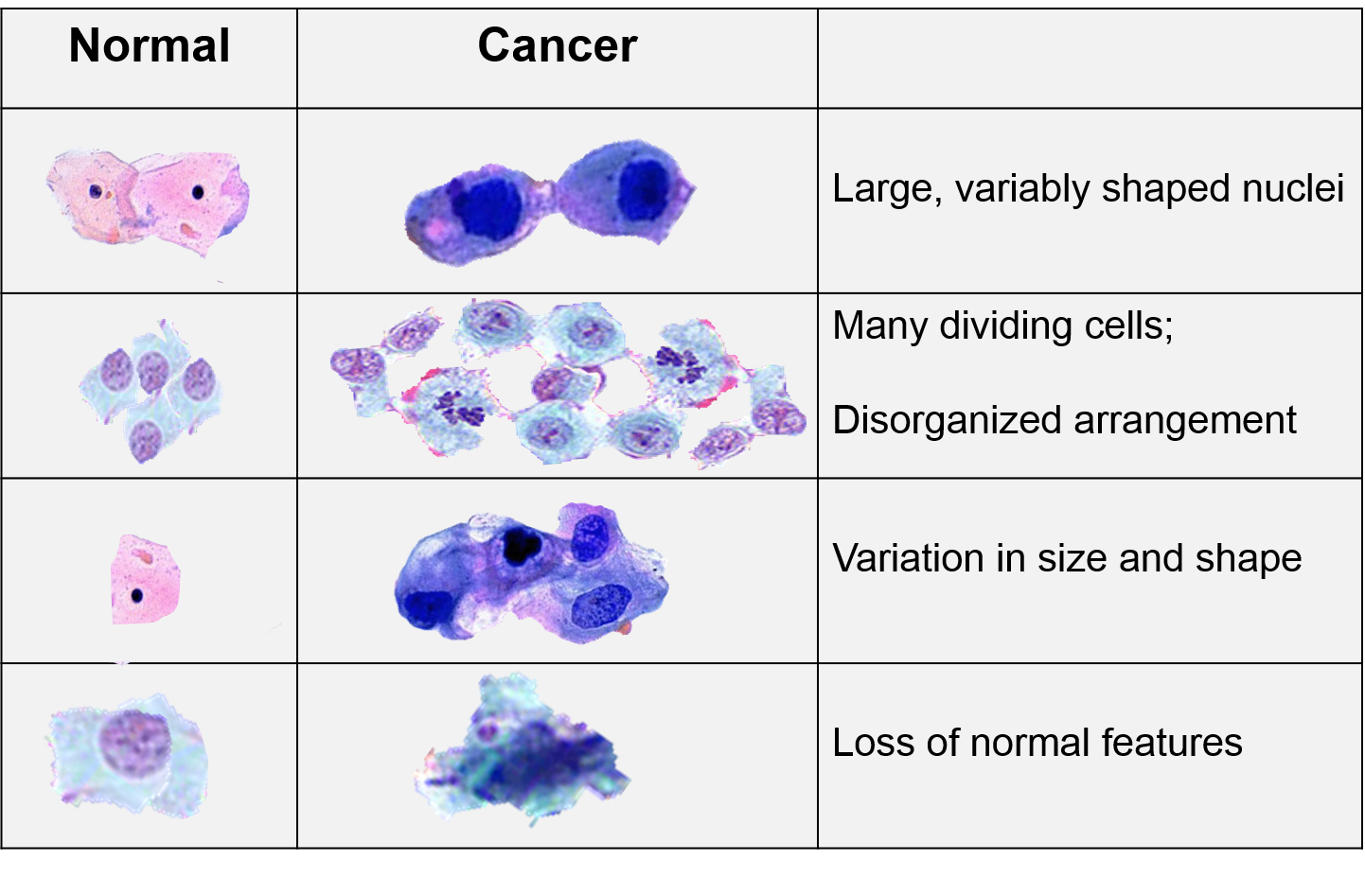
Suppose that oncologists…
…wish to build a model to predict which tumors are malignant (versus benign) based on the visual characteristics of the cells:
- Average radius of all visible cell nuclei (
Radius) - Indicator of whether the visible cell nuclei are misshapen and concave (
Concave)
Logistic models
The linear predictor component of a GLM is the same in logistic and linear regression:
- Additive models incorporate information from both visual characteristics simultaneously
- Interaction models allow the relationship between the radius of the nuclei and tumor malignancy to differ depending on concavity (and for the nuclei shape/tumor malignancy relationship to differ depending on the radius!)
Additive model for tumor malignancy
Let \(\pi\) be the probability that a particular tumor is malignant
Population Model
\[ \text{logit}\left\{\pi(\texttt{Radius}, \texttt{Concave})\right\} = \beta_0 + \beta_1(\texttt{Radius}) + \beta_2(\texttt{Concave}) \]
Fitted Model
\[ \text{logit}\left\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave})\right\} = -13.132 + 2.718(\texttt{Radius}) + 3.319(\texttt{Concave}) \]
Fitting the additive model
Visualizing the additive model
Additive Model
\[\begin{align*} \text{Convex Cells:} &\; \text{logit}\{\widehat{\pi}(\texttt{Radius},\texttt{Concave}= 0)\} = -13.132 + 2.718(\texttt{Radius}) \\ \text{Concave Cells:} &\; \text{logit}\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave} = 1)\} = -9.813 + 2.718(\texttt{Radius}) \end{align*}\]
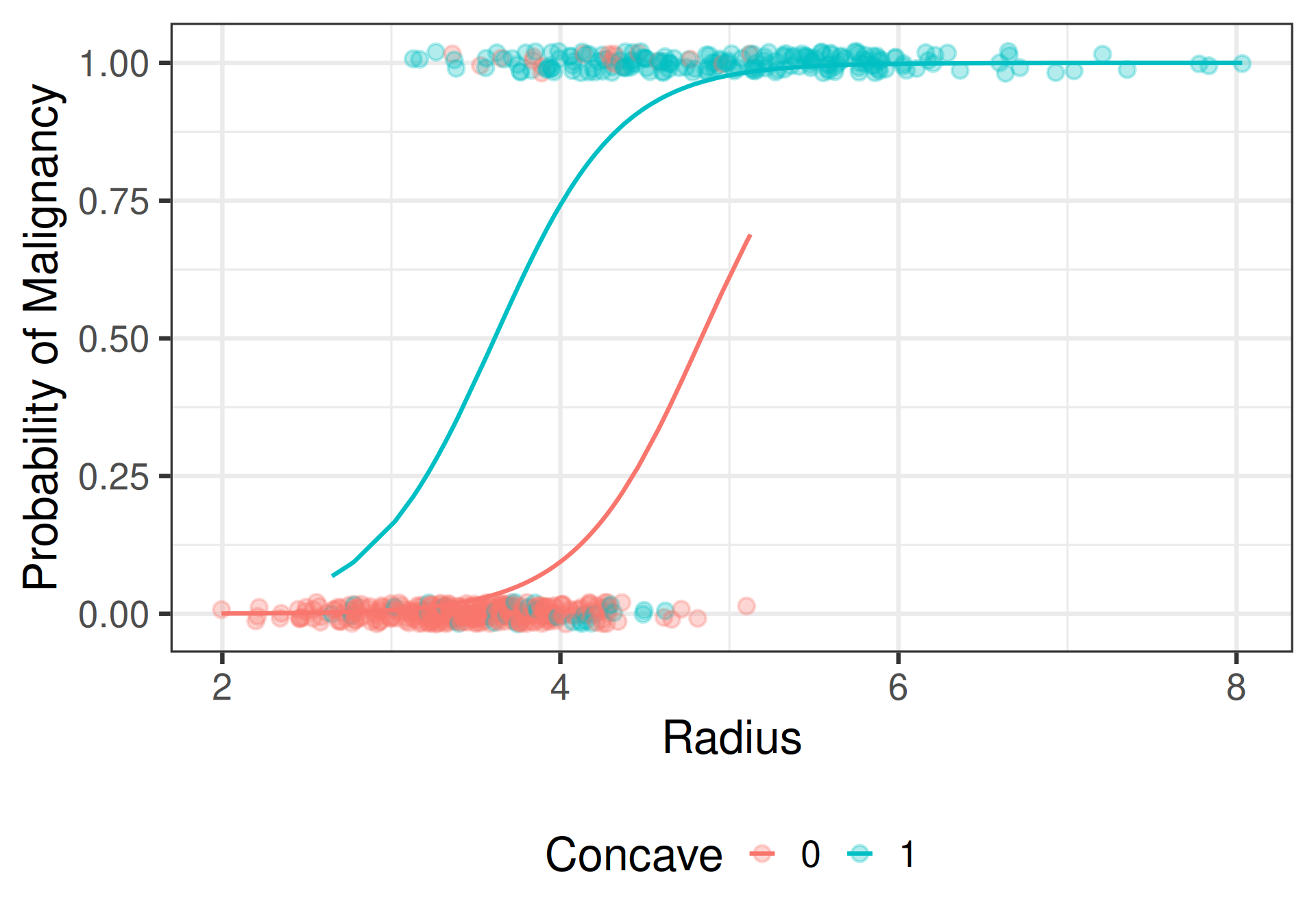
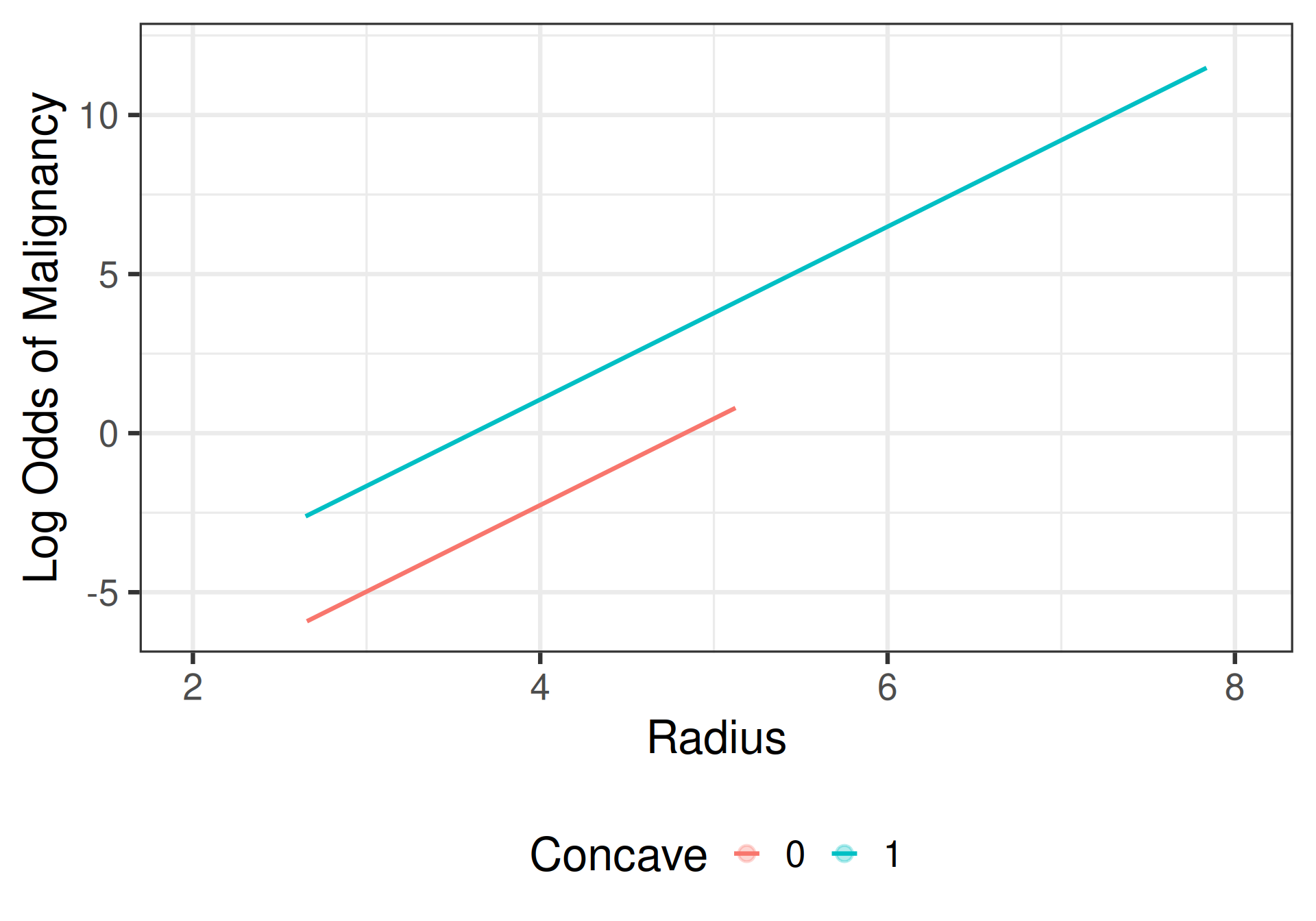
Interpretation: additive model
Additive model
\[ \text{logit}\left\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave})\right\} = -13.132 + 2.718(\texttt{Radius}) + 3.319(\texttt{Concave}) \]
- Intercept: The estimated odds of malignancy among tumors with an average cell nuclei radius of 0 \(\mu\)m and convex nuclei is 0.000002.
\[\text{Scratch work: }e^{-13.132}\approx 0.000002\]
Interpretation: additive slopes
- Radius: Holding cell shape constant, each additional \(\mu\)m increase in the average radius of the cell nuclei is associated with a 15.15 times change (1,415% increase) in the odds of tumor malignancy.
\[\text{Scratch work: }e^{2.718} = 15.15 \implies (15.15-1)\times 100\% = 1415\%\]
- Concave: Among tumors with the same average cell radius, the odds that a tumor with concave cell nuclei is malignant are 27.63 times as great as (2,663% greater than) the odds when the cells are convex.
\[\text{Scratch work: }e^{3.319} = 27.63 \implies (27.63-1)\times 100\% = 2663\%\]
Interaction model for malignancy
If we want to allow the relationship between average cell radius (or cell shape) and \(\pi\) to differ depending on the other visual characteristic…
Population Model
\[\begin{align*} \text{logit}\{\pi(\texttt{Radius}, \texttt{Concave})\} = & \ \beta_0 + \beta_1(\texttt{Radius}) + \beta_2(\texttt{Concave}) \\ &+ \beta_3(\texttt{Radius})(\texttt{Concave}) \end{align*}\]
Fitted Model
\[\begin{align*} \text{logit}\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave})\} = & \ -15.036 + 3.189(\texttt{Radius}) + 6.455(\texttt{Concave}) \\ &- 0.778(\texttt{Radius})(\texttt{Concave}) \end{align*}\]
Fitting the interaction model
Call:
glm(formula = Malignant ~ Radius * Concave, family = "binomial",
data = cell)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -15.0357 2.4698 -6.088 1.14e-09 ***
Radius 3.1889 0.6050 5.271 1.36e-07 ***
Concave 6.4551 3.0460 2.119 0.0341 *
Radius:Concave -0.7784 0.7472 -1.042 0.2976
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 751.44 on 568 degrees of freedom
Residual deviance: 222.91 on 565 degrees of freedom
AIC: 230.91
Number of Fisher Scoring iterations: 7Visualizing interaction model
Interaction Model
\[\begin{align*} \text{Convex Cells:} &\; \text{logit}\{\widehat{\pi}(\texttt{Radius},\texttt{Concave} = 0)\} = -15.036 + 3.189(\texttt{Radius}) \\ \text{Concave Cells:} &\; \text{logit}\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave} = 1)\} = -8.581 + 2.411(\texttt{Radius}) \end{align*}\]
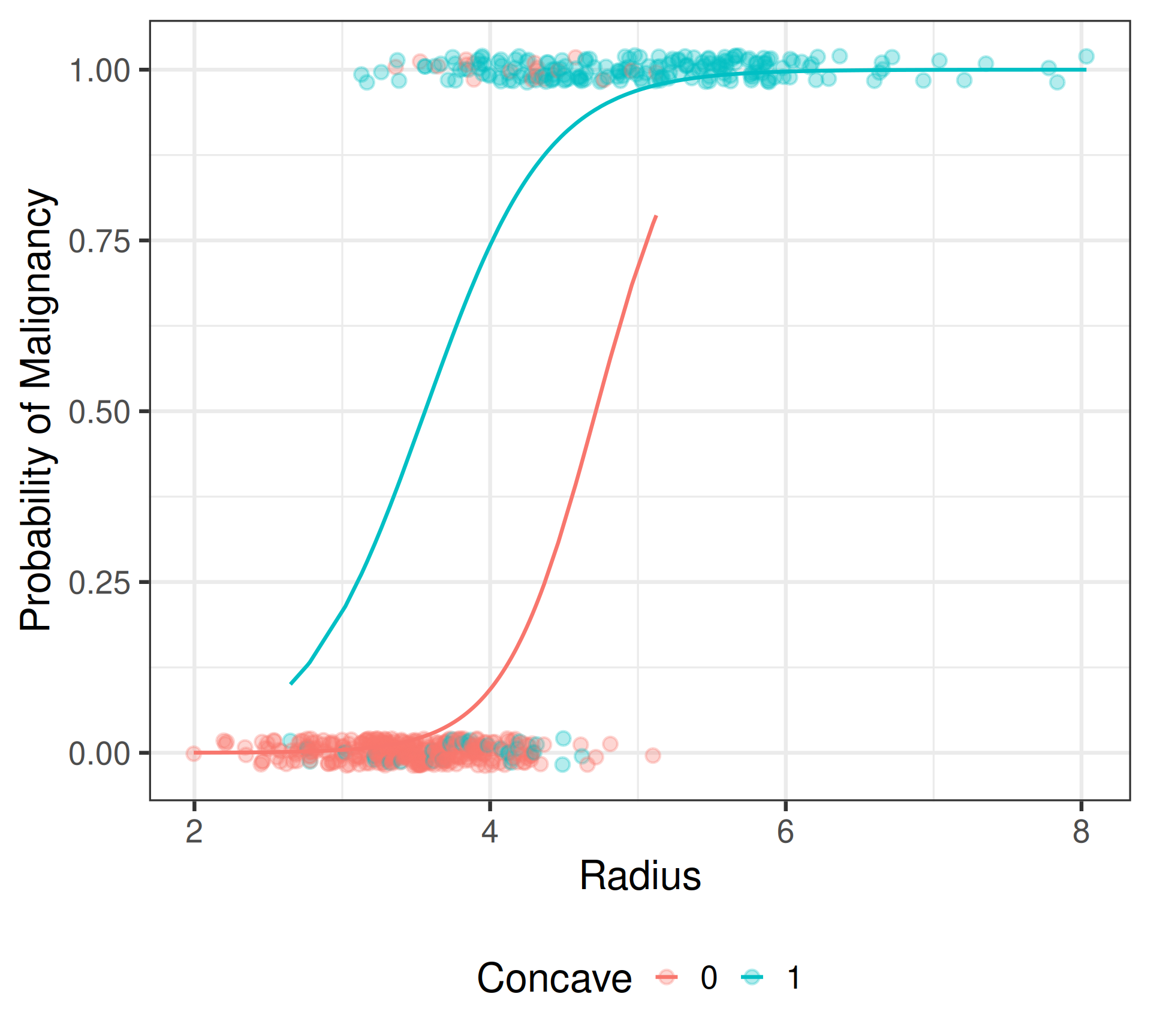
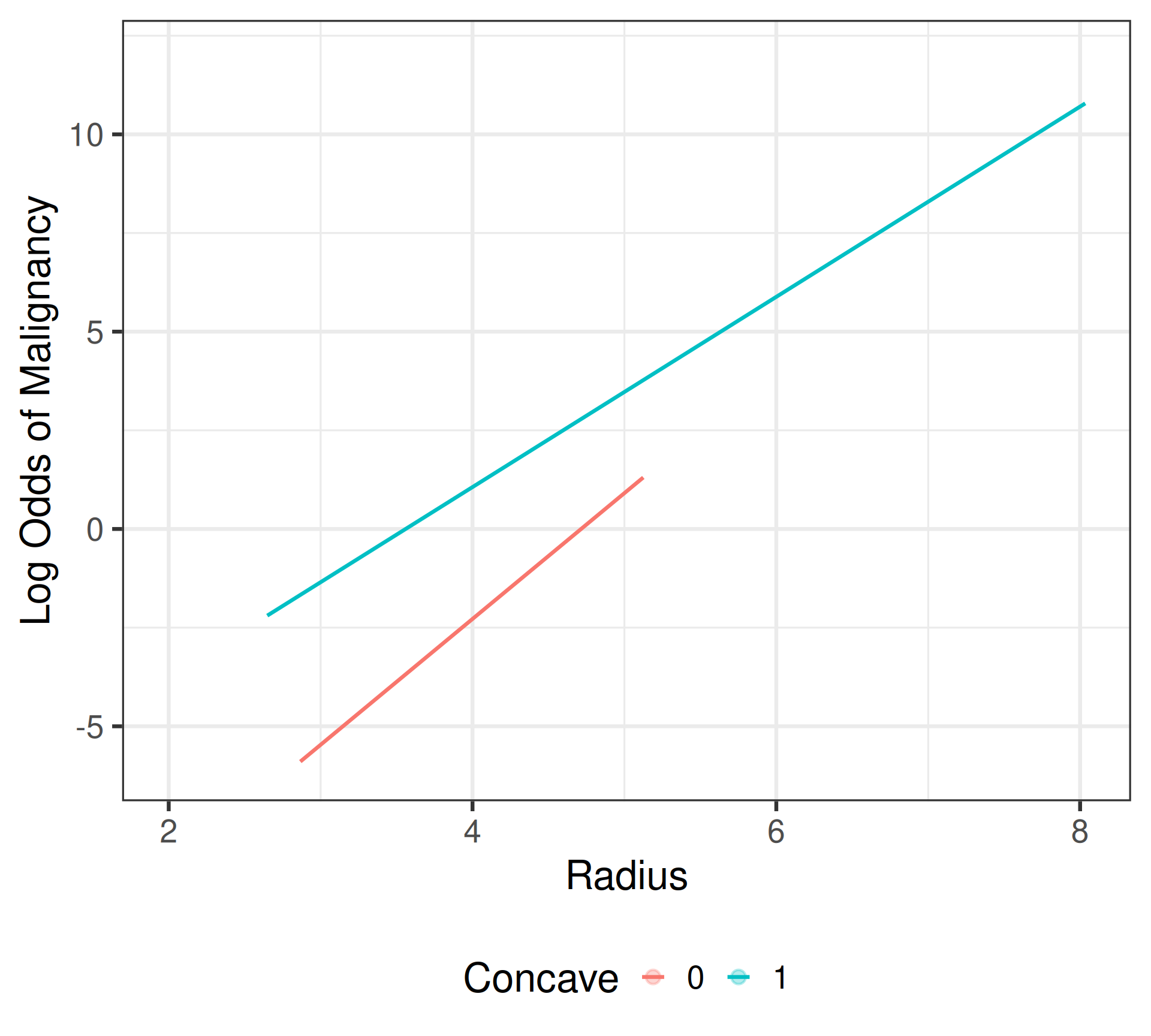
Interpretation: Radial size
Our interpretations now require us to specify at which value we are holding the other variable involved in the interaction constant:
Interaction Model
\[\begin{align*} \text{logit}\{\widehat{\pi}(\texttt{Radius}, \texttt{Concave})\} = & \ -15.036 + 3.189(\texttt{Radius}) + 6.455(\texttt{Concave}) \\ &- 0.778(\texttt{Radius})(\texttt{Concave}) \end{align*}\]
- Radius: For tumors with convex cell nuclei, each one \(\mu\)m increase in the average nuclei radius is associated with a 24.26 times change (2,326% increase) in the odds of tumor malignancy. \[\text{Scratch work: }e^{3.189} = 24.26 \implies (24.26-1)\times 100\% = 2326\%\]
- Radius: For tumors with concave cell nuclei, each one \(\mu\)m increase in the average nuclei radius is associated with a 11.15 times change (1,015% increase) in the odds of tumor malignancy. \[\text{Scratch work: }e^{3.189 - 0.778} = 11.15 \implies (11.15-1)\times 100\% = 1015\%\]
Interpretation: Concavity
- Concave: For tumors with an average nuclei radius of 4 \(\mu\)m, the odds of malignancy are 28.30 times as high (2,730% higher) when the cell nuclei are concave as they are when the cell nuclei are convex. \[\text{Scratch work: }e^{6.455 - 0.778(4)} = 28.30 \implies (28.30-1)\times 100\% = 2730\%\]
- Concave: For tumors with an average nuclei radius of 7 \(\mu\)m, the odds of malignancy are 2.74 times as high (174% higher) when the cell nuclei are concave as they are when the cell nuclei are convex. \[\text{Scratch work: }e^{6.455 - 0.778(7)} = 2.74 \implies (2,74-1)\times 100\% = 174\%\]
Model fit
# A tibble: 2 × 8
null.deviance df.null logLik AIC BIC deviance df.residual nobs
<dbl> <int> <dbl> <dbl> <dbl> <dbl> <int> <int>
1 751. 568 -112. 230. 243. 224. 566 569
2 751. 568 -111. 231. 248. 223. 565 569Classification
Related questions
- Prediction: What is the probability that a person with an average cell radius of 5\(\mu\)m and convex cell nuclei has a malignant tumor?
- Classification: Can we use these probabilities to determine who actually does and does not have a malignant tumor?
Calculating Predicted Probabilities
Research Question
What is the probability that a person with an average cell radius of 5 \(\mu\)m and convex cell nuclei has a malignant tumor?
\[\widehat{\pi}(5, 0) = \frac{e^{-13.132 + 2.718(5) + 3.319(0)}}{1 + e^{-13.132 + 2.718(5) + 3.319(0)}} = 0.612\]
CIs for Predicted Odds
Research Question
What is the probability that a person with an average cell radius of 5 \(\mu\)m and convex cell nuclei has a malignant tumor?
\[\begin{align*} \text{logit}(\hat{\pi}) &\pm z^* \cdot SE_{\text{logit}(\hat{\pi})} \\ 0.456 &\pm 1.96 \cdot 0.442 = (-0.410, 1.322) \end{align*}\]
CIs for Predicted Probabilities
- First build them for the log odds: \[ 0.456 \pm (1.96)(0.442) = (-0.410, 1.322) \]
- Then transform both endpoints back to probability scale: \[ \left(\frac{e^{-0.410}}{1 + e^{-0.410}}, \ \frac{e^{1.322}}{1 + e^{1.322}}\right) = (0.399, 0.790) \]
Answers
- Prediction: What is the probability that a person with an average cell radius of 5 \(\mu\)m and convex cell nuclei has a malignant tumor?
- We estimate this probability to be 61.2%.
- We are 95% confident that the true probability that an individual with an average cell radius of 5 \(\mu\)m and convex cell nuclei has a malignant tumor is between 39.9% and 70.0%.
- Classification: Can we use these probabilities to determine who actually does and does not have malignant cancer?
- How well did our model do at making these predictions?
- If all we had to go off of was the predicted probability \(\widehat{\pi}\), could we reliably figure out who actually has a malignant tumor?
Classification with logistic regression
Intuition
Model does a good job at predicting tumor malignancy if it assigns higher values of \(\widehat{\pi}\) to people who actually have malignant tumors and lower values of \(\widehat{\pi}\) to people who do not
- A new patient comes along whose true tumor status we don’t know, but whose predicted probability is \(\widehat{\pi} = 0.612\)
- Should we refer this patient for surgery?
Consider two models
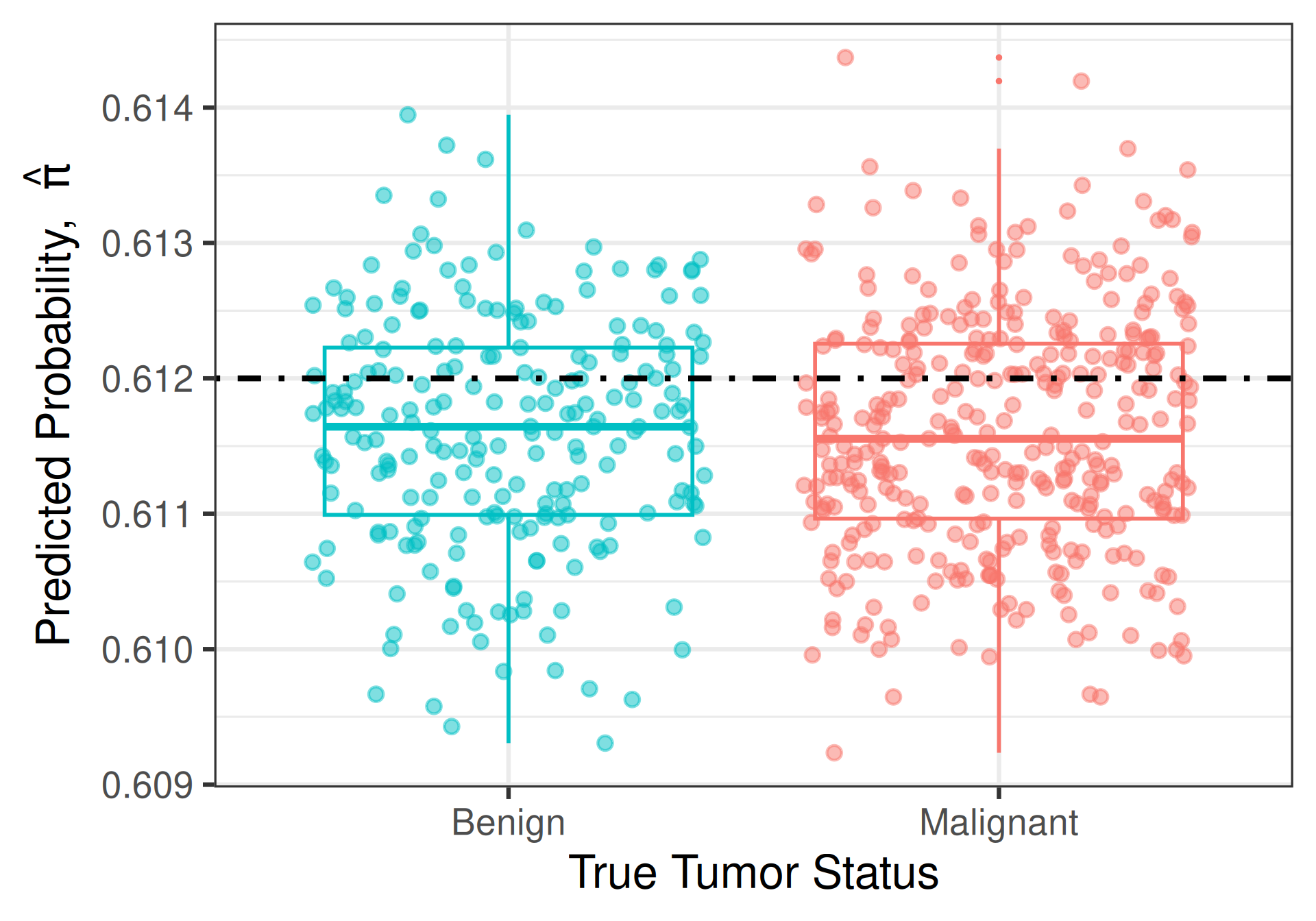
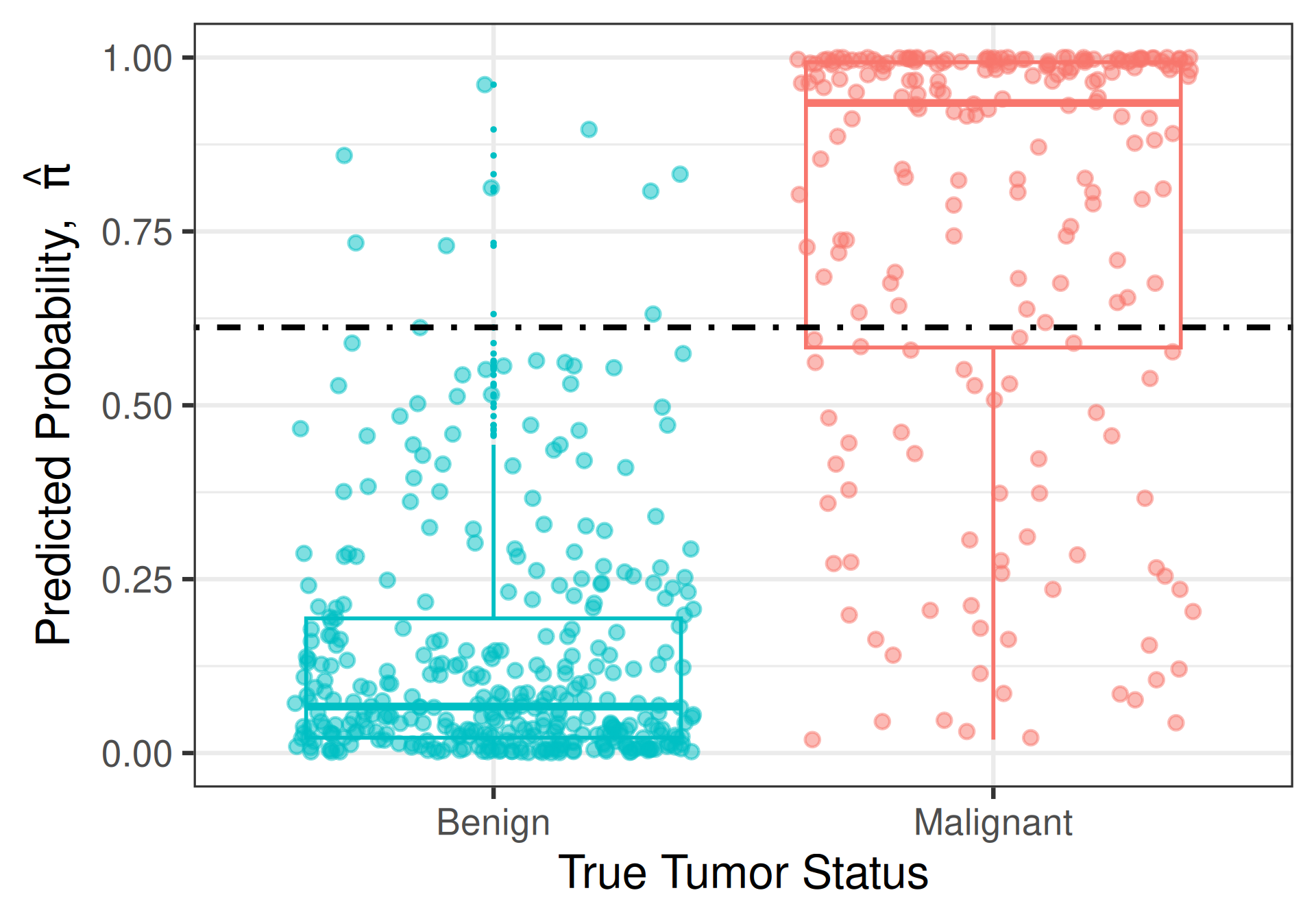
Classification Cut-Offs
- Set a cut-off: \(\pi_0 = 0.5\)
\[\begin{alignat*}{2} &\widehat{\pi} > 0.5 \quad &&\implies \quad \text{classify as a "malignant" tumor, }\widehat{y} = 1 \\ &\widehat{\pi} \leq 0.5 \quad &&\implies \quad \text{classify as a "benign" tumor, }\widehat{y} = 0 \\ \end{alignat*}\]
# A tibble: 4 × 3
.fitted pred Malignant
<dbl> <fct> <dbl>
1 0.0125 Benign 0
2 0.0265 Benign 0
3 0.0276 Benign 0
4 0.0173 Benign 0Classification tables
Any classification is either correct or incorrect:
pred
Malignant Benign Malignant Sum
0 335 22 357
1 24 188 212
Sum 359 210 569Tip
Believe it or not, these tables are called confusion matrices!
Classification metrics
- Sensitivity: the proportion of true successes that we correctly classify as a “success” \[\begin{equation*} \text{sens} = \frac{\#\text{ correctly predicted malignant}}{\#\text{ with malignant tumors}} = \frac{TP}{TP + FN} \end{equation*}\]
- Specificity: the proportion of true failures that we correctly classify as a “failure” \[\begin{equation*} \text{spec} = \frac{\#\text{ correctly predicted benign}}{\#\text{ with benign tumors}} = \frac{TN}{FP + TN} \end{equation*}\]
Try \(\pi_0 = 0.5\)
# A tibble: 3 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 accuracy binary 0.919
2 sens binary 0.938
3 spec binary 0.887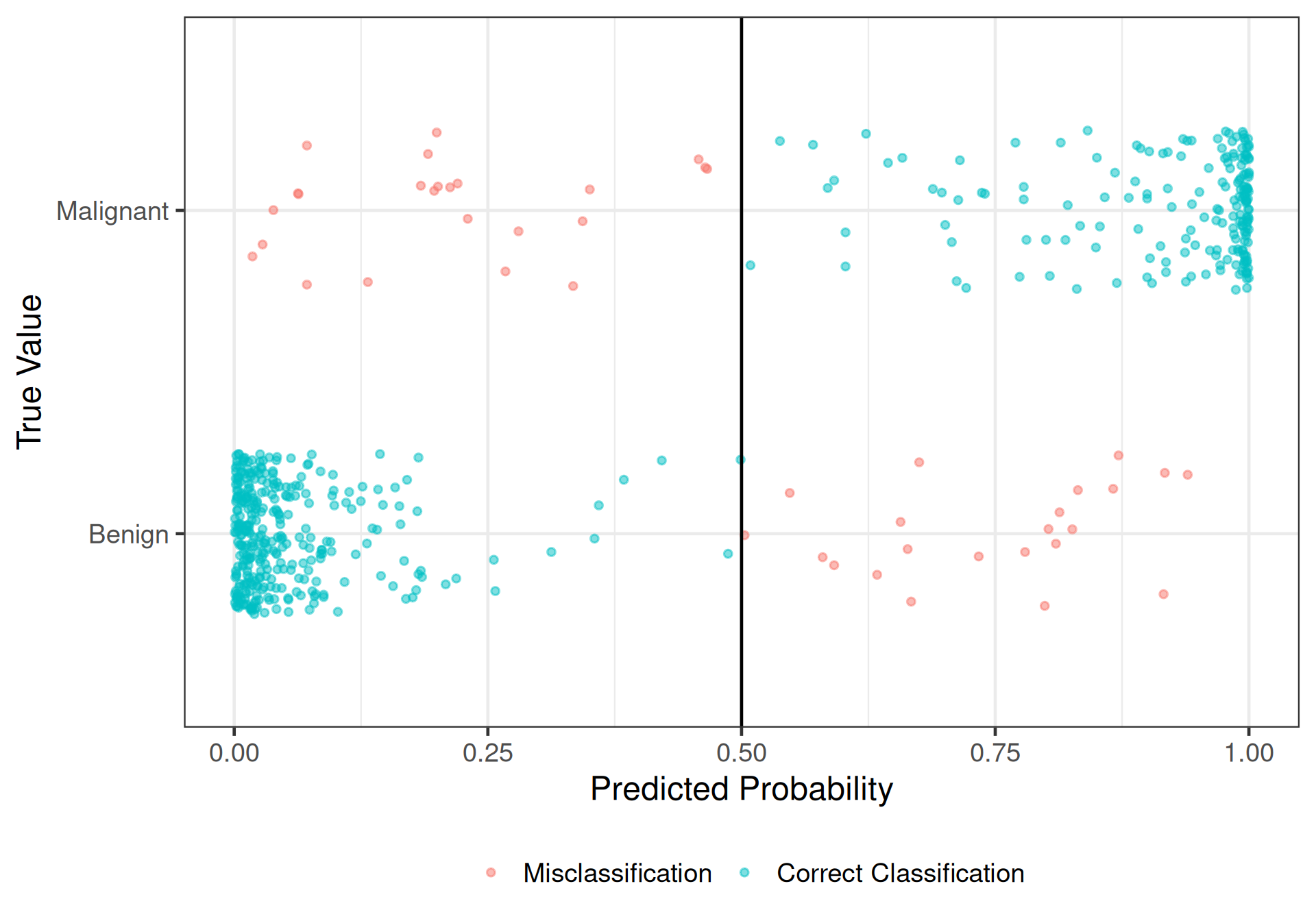
Try \(\pi_0 = 0.05\)
# A tibble: 3 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 accuracy binary 0.789
2 sens binary 0.672
3 spec binary 0.986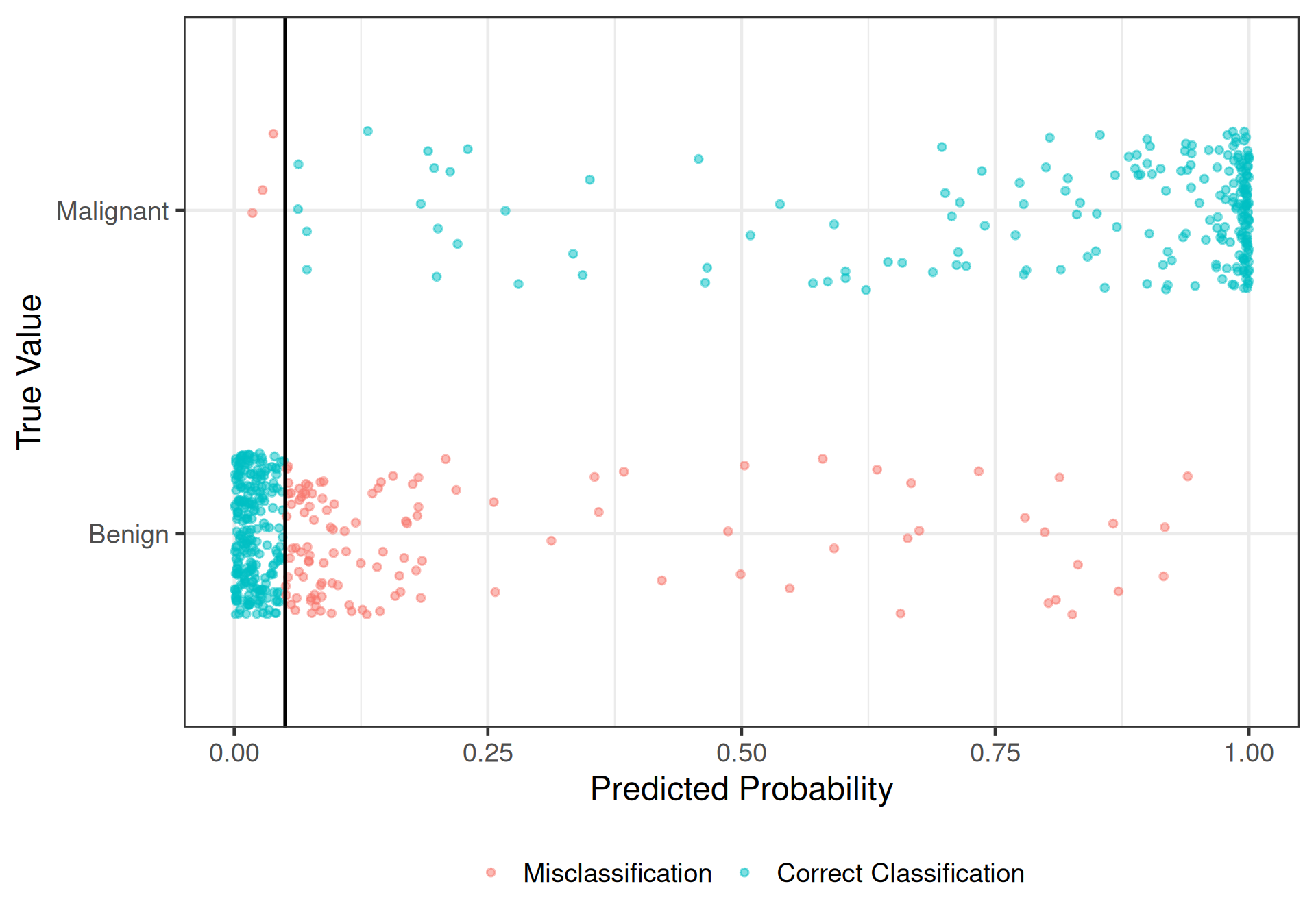
Try \(\pi_0 = 0.9\)
# A tibble: 3 × 3
.metric .estimator .estimate
<chr> <chr> <dbl>
1 accuracy binary 0.870
2 sens binary 0.992
3 spec binary 0.665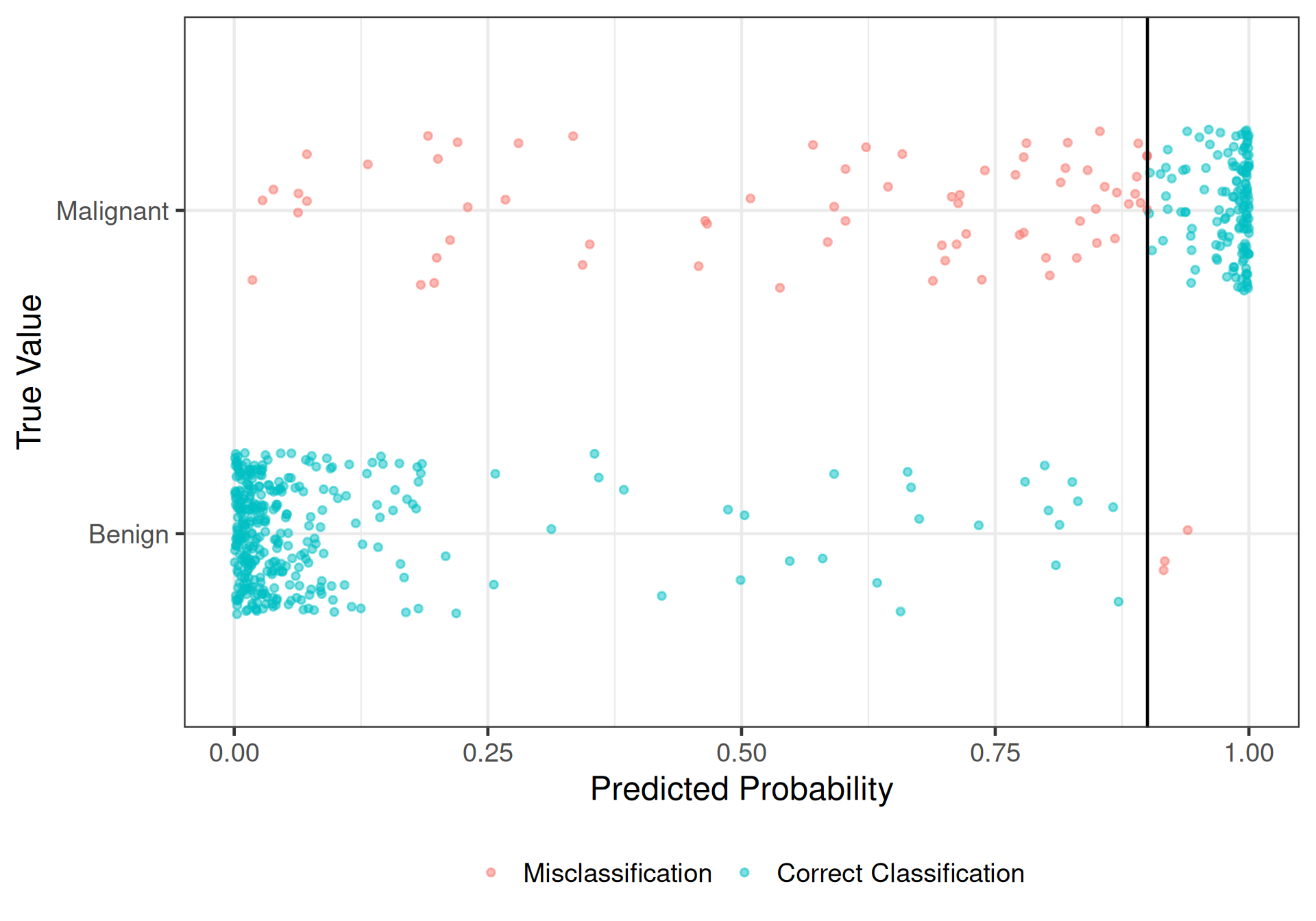
Trade-offs
- A model with high discrimination makes it easier to find a cut-off that produces a good balance between sensitivity and specificity

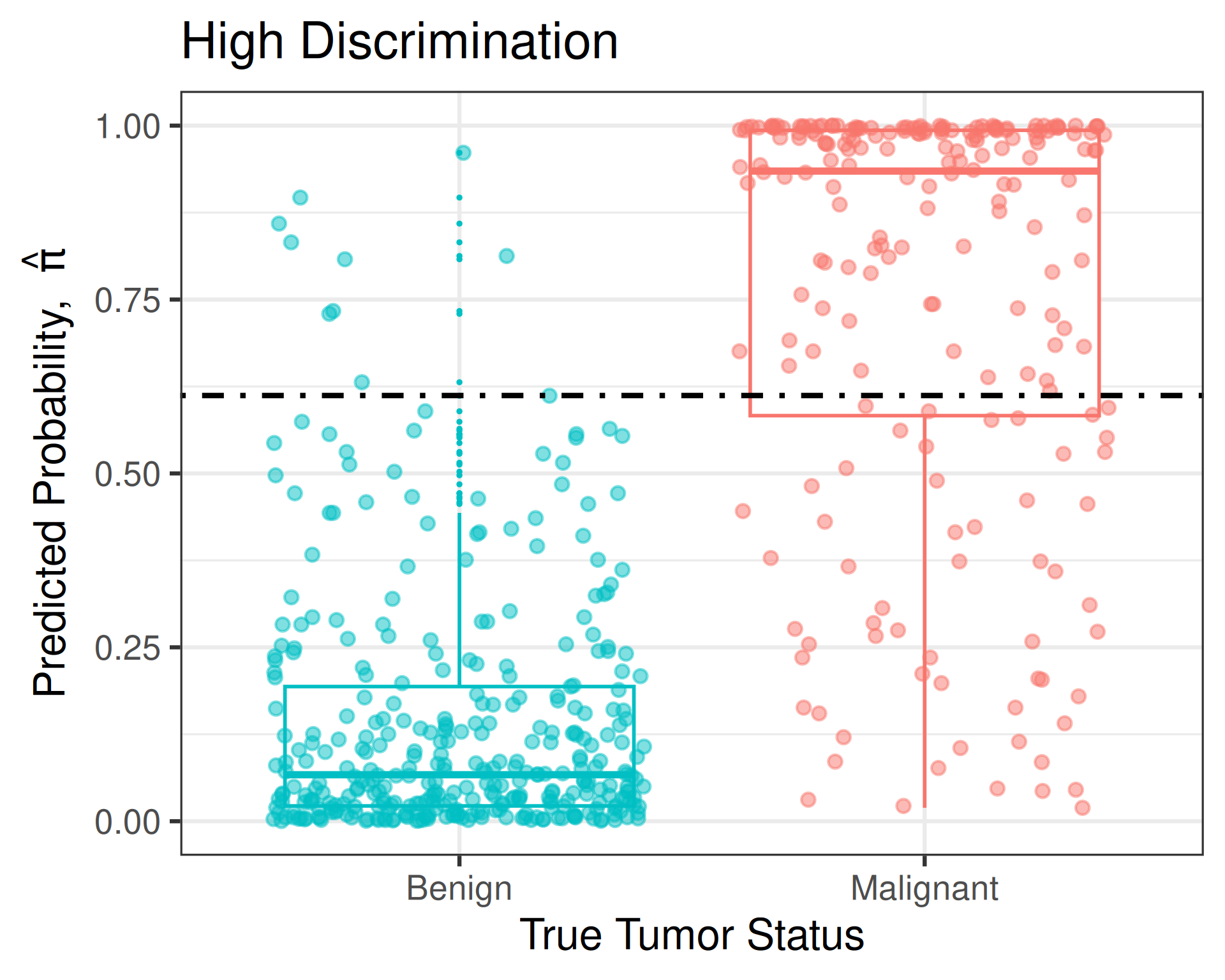
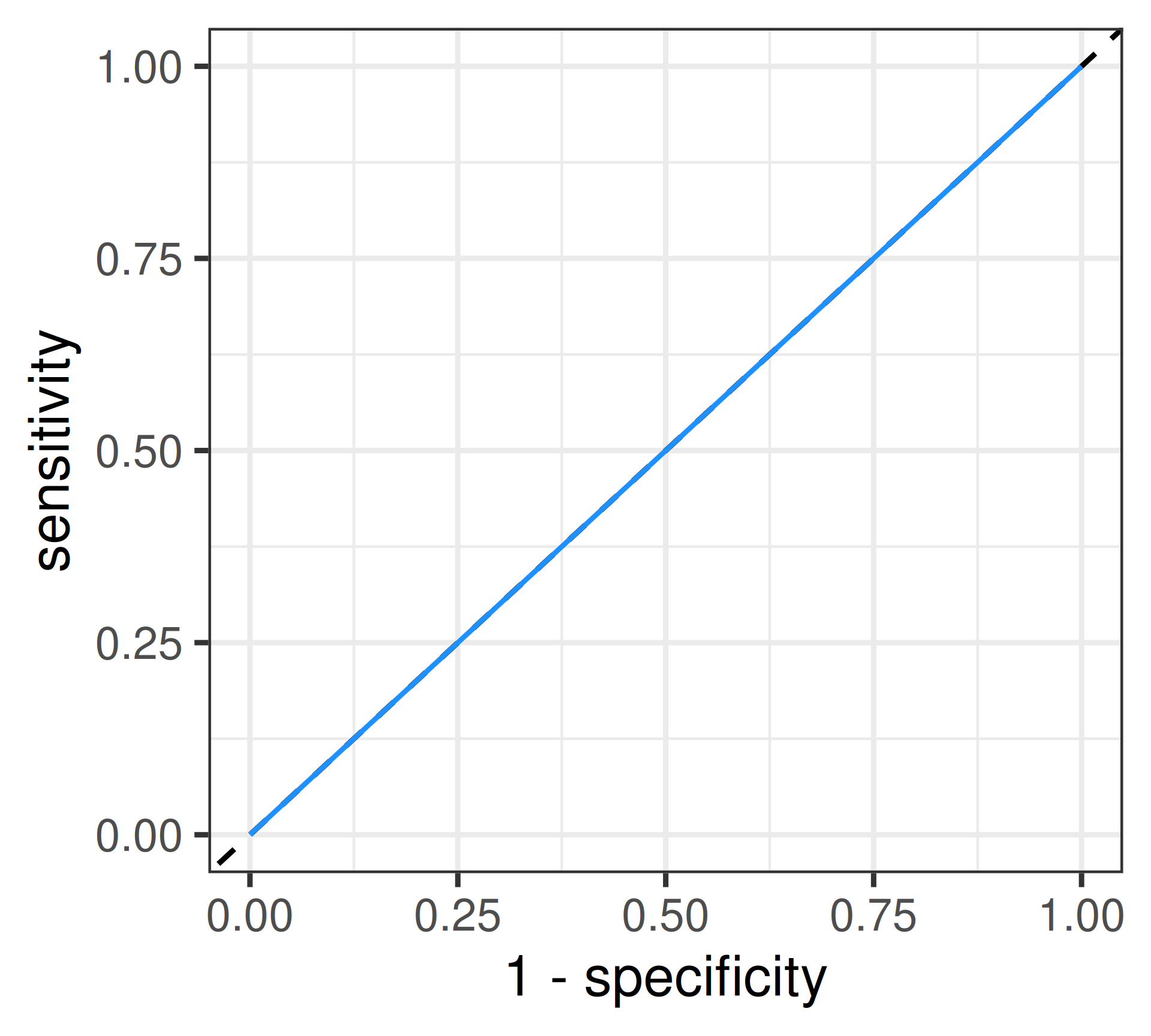
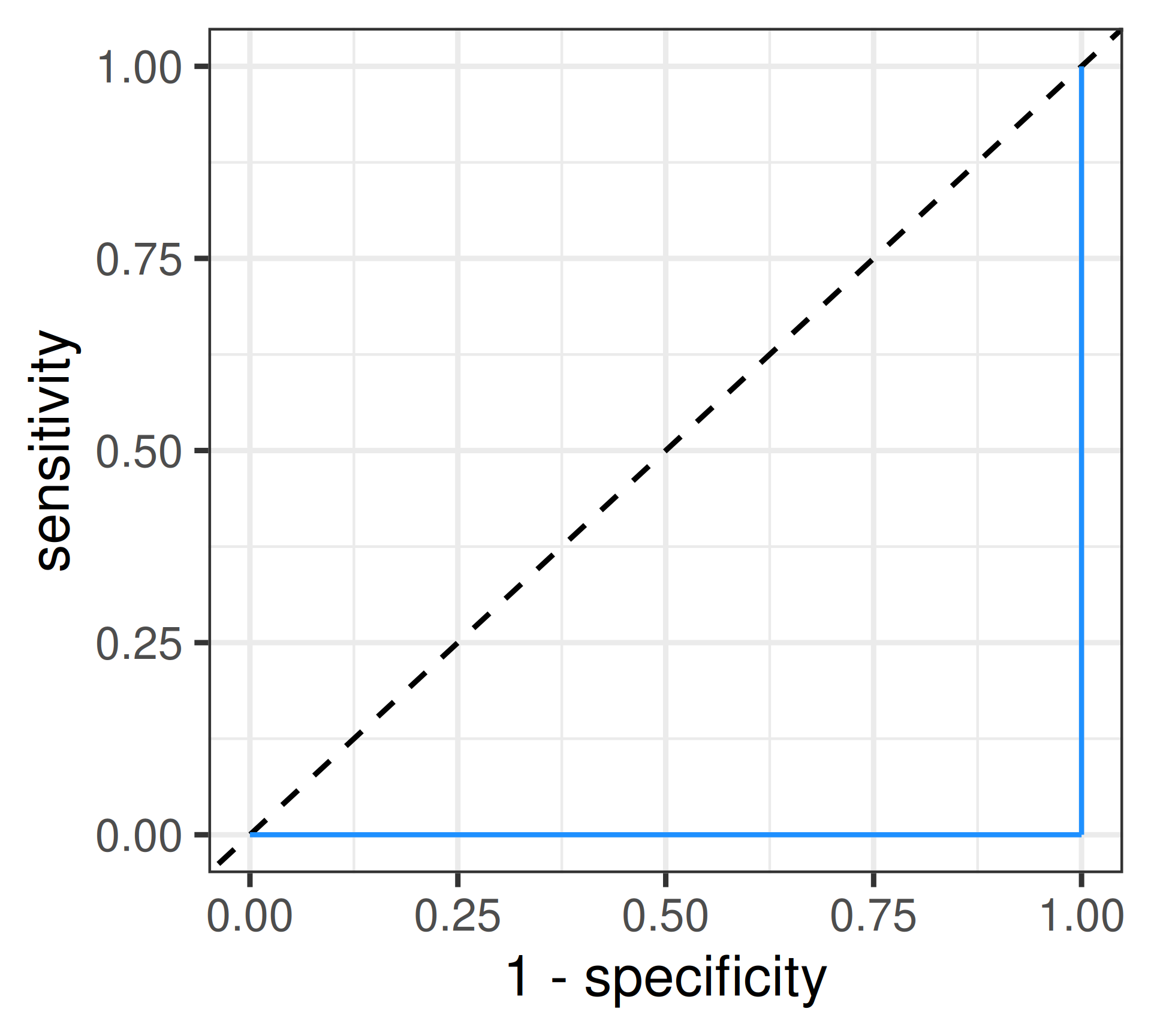
ROC curve
Receiver Operating Characteristic: Visualizes the trade-off between sensitivity and specificity across a range of cut-offs
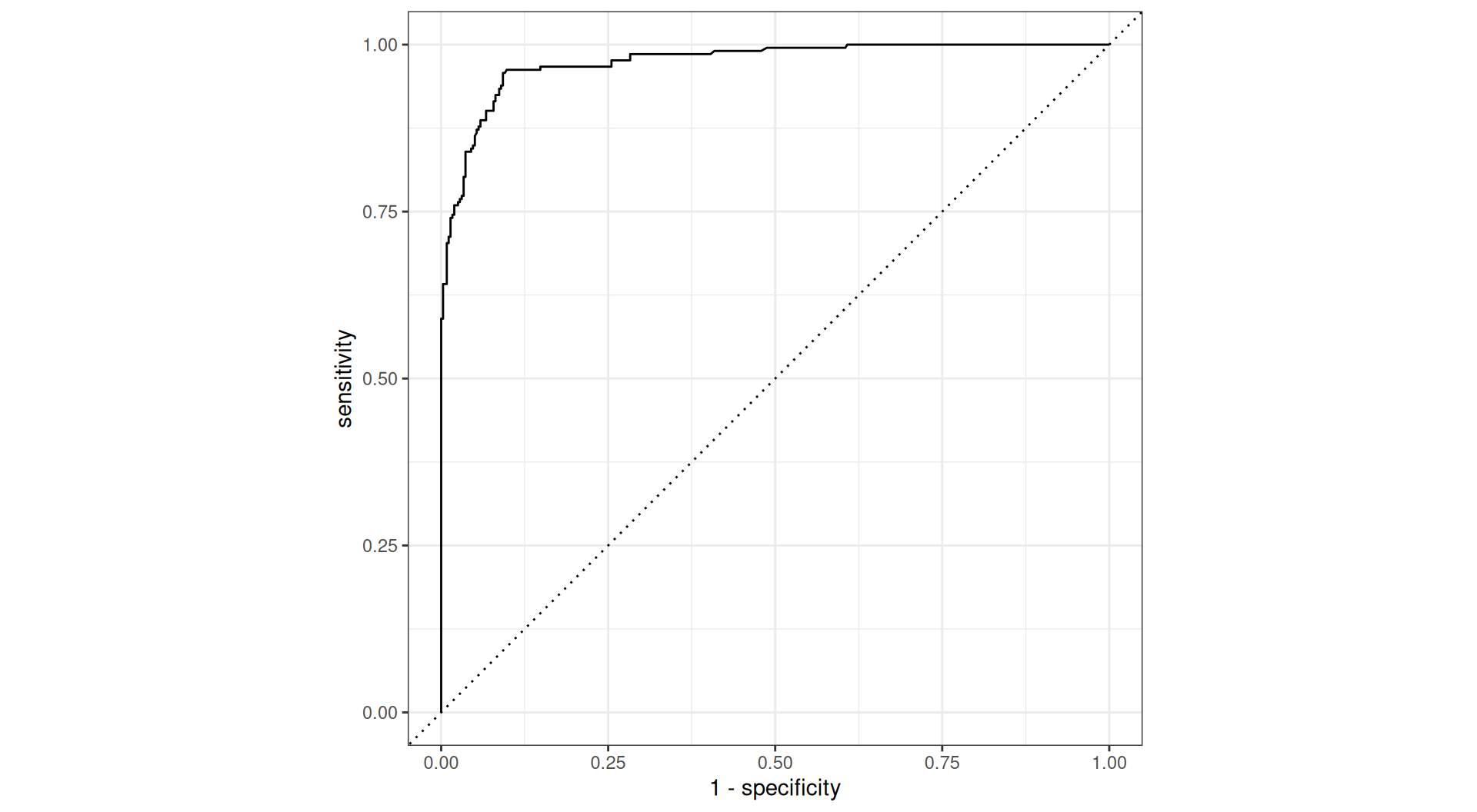
Area under the ROC curve (AUC)
- single number summarizing the overall discrimination
- a.k.a., the c-statistic or concordance index
- If we randomly select one success and one failure, what are the chances that the true success has a higher predicted probability than the true failure?
Note
- Would you prefer a larger or smaller AUC?
- What would the AUC be if the ROC curve fell exactly on the \(y=x\) line?
Extreme ROC curves
Perfect Discrim.: \(AUC = 1\)
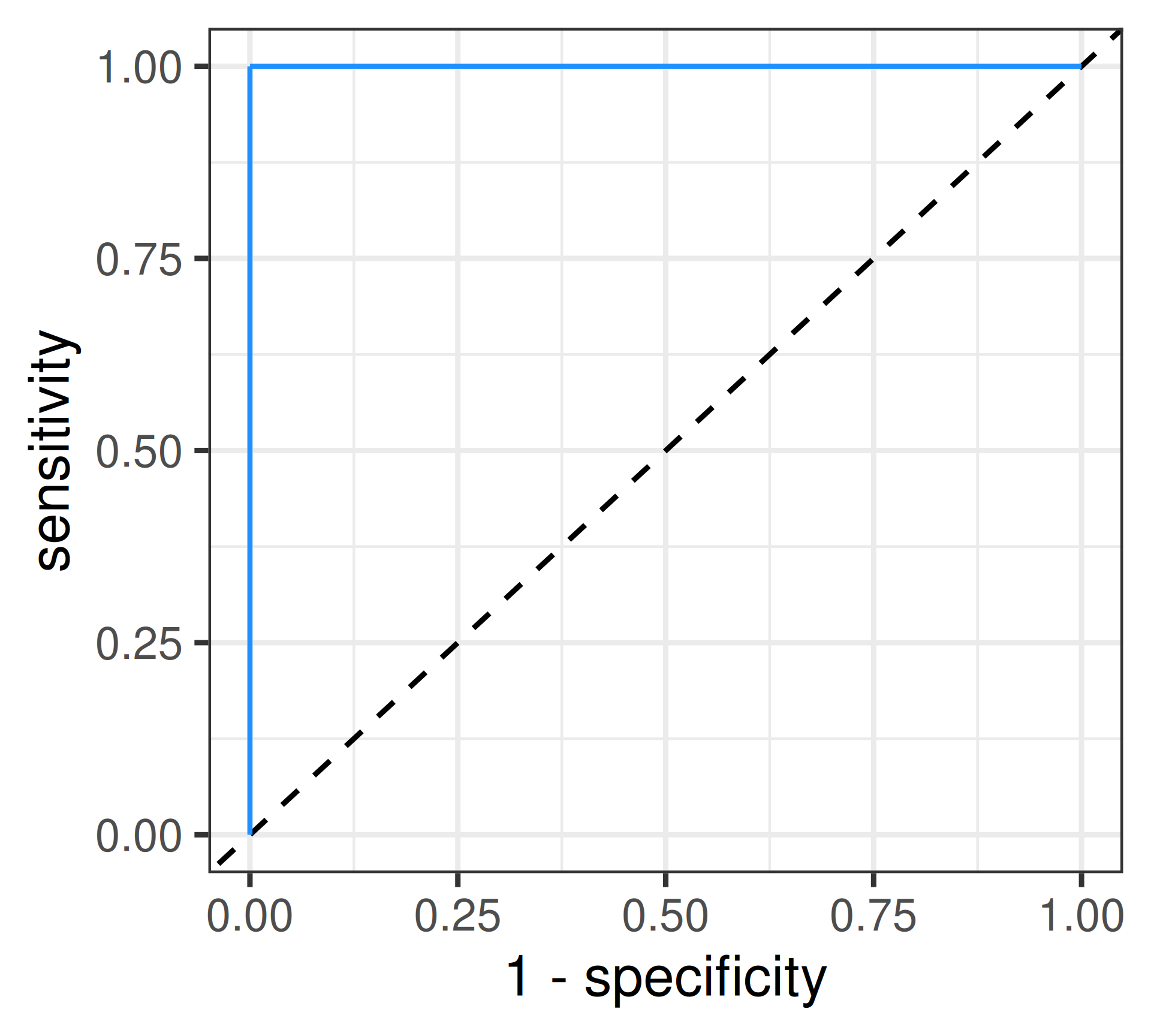
Random Guessing: \(AUC = 0.5\)
Data Coding Error?: \(AUC = 0\)
Selecting a classification cut-off (\(\pi_0\))
- Different choices of \(\pi_0\) will navigate the trade-off between sensitivity and specificity differently
- Desirable \(\pi_0\) is thus context-dependent:
- If we want to minimize the chance we miss actual malignant tumors, we want high sensitivity \(\iff\) select a lower cut-off for \(\pi_0\)
- If we want to minimize the chance we mistakenly diagnose benign tumors as malignant, we want high specificity \(\iff\) select a higher cut-off for \(\pi_0\)
- If we value both metrics equally, another option is to maximize the vertical distance to the reference \(y = x\) line (called Youden’s index): \[\max\left\{\text{sensitivity} + \text{specificity} - 1\right\}\]
![]()
SDS 291